9. Mai 2025
Große Proteinstrukturen besser beschreiben
Präziseres Modell stellt Flexibilität und Eigenschaften von Proteinen dar
Mit einer deutlich verbesserten Methode optimiert eine Gruppe um FIAS-Fellow Sebastian Thallmair die Berechnung von Proteinstrukturen. Dem internationalen Team ist es gelungen, die Flexibilität und mechanischen Eigenschaften von Proteinen präziser zu beschreiben.
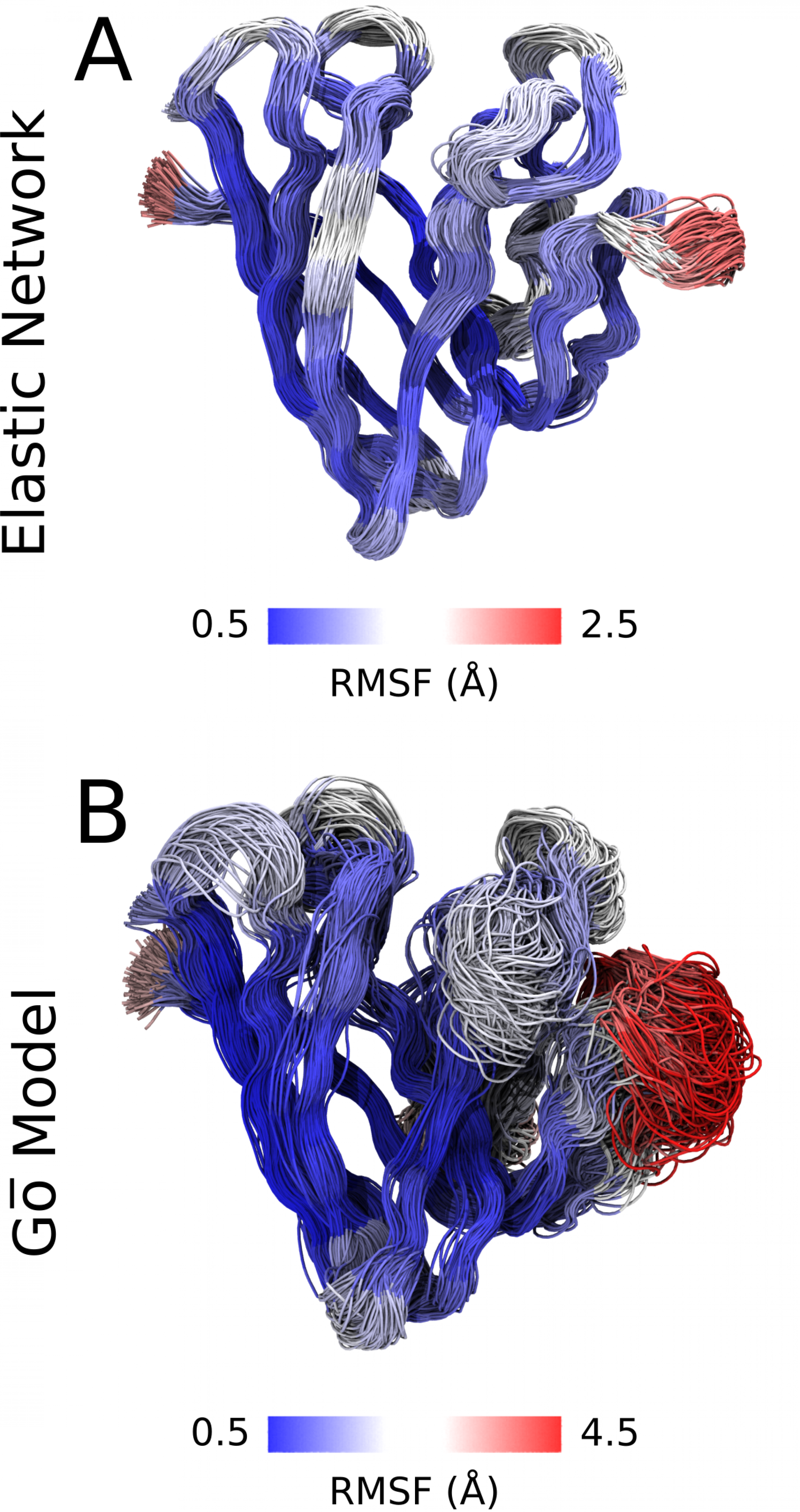
Die Struktur größerer Proteine oder gar Proteinkomplexe Atom für Atom zu berechnen ist kompliziert und erfordert hohen Rechenaufwand – „das kann Jahre dauern“, erklärt Sebastian Thallmair, Fellow am FIAS. Einem großen internationalen Team ist es nun gelungen, diese Berechnungen zu verbessern. Dabei verwenden sie eine Methode, die auch den Rechenaufwand verringert. Sie stellen nicht jedes Atom einzeln dar, sondern fassen diese in kleinen Gruppen zusammen. Dies ist entscheidend für den geringeren Rechenaufwand. Da jedoch „kleinere“ Wechselwirkungen wie Wasserstoffbrücken-Bindungen Proteinstrukturen entscheidend stabilisieren, müssen diese teilweise wieder eingeführt werden. Das Team hat eine deutlich verbesserte Methode entwickelt, die Proteinmodelle stabilisiert, ihnen jedoch gleichzeitig eine hohe Flexibilität ermöglicht.
„Durch die Gruppierung von Atomen rechnen wir mit einem Zehntel bis Zwanzigstel der Teilchen und müssen in unserem optimierten Modell nur hundert bis tausend zusätzliche Bindungen berücksichtigen, um die Flexibilität der Proteine realistisch zu beschreiben“, erklärt Thallmair. „Darüber hinaus erlaubt uns die vielseitige Methode auch, die Wechselwirkung mit der Umgebung zu verfeinern“. Das Modell ist damit deutlich effizienter als Berechnungen von Proteinen mit atomistischer Auflösung und lässt sich dank Verbesserungen auf entsprechenden Supercomputern, wie sie dem FIAS zu Verfügung stehen, verwenden. Das weit verbreitete Martini-Modell zur Berechnung von Proteinen wird damit breiter anwendbar und gibt zudem die Proteindynamik deutlich besser wieder.
An verschiedenen Beispielen weisen Thallmair und sein Team diese Optimierungen nach:
- An Proteinen, die in der Membran von Zellen wichtige Vermittlungsaufgaben zwischen Innen und Außen übernehmen, zeigen sie, dass ihr Modell deren Struktur mindestens vergleichbar gut darstellt und gleichzeitig die Elastizität der Proteine – ihre Beweglichkeit abhängig von äußeren Einflüssen - besser beschreibt.
- Denselben Effekt zeigen sie für Proteine, die andere Moleküle binden, etwa Medikamente: Die verbesserte Flexibilität der Proteine erlaubt es beispielsweise genauer zu beschreiben, wie sich Proteintaschen bei der Bindung eines Moleküls anpassen.
- Die verbesserten mechanischen Eigenschaften werden bei einem Protein deutlich, dessen Mutation in lediglich einer Aminosäure eine schwere Erkrankung des Nervensystems hervorruft (amyotrophe Lateralsklerose). Die Änderung führt dazu, dass das aktive Zentrum weiter aufgeht und das Protein so seine Funktion verliert. Für diese Beobachtung war es entscheidend, dass die Forschenden dank des verringerten Rechenaufwands eine Simulationsdauer von einer Millisekunde erreichen konnten.
- Anhand von Mutationen im Spike-Protein des Corona-Virus zeigten die Forschenden, dass die Methode auch beschreiben kann, wie die Mutationen die Wechselwirkungen zwischen Wirt und Virus verbessern.
Mit der großen Bandbreite an Beispielen zeigt das Team, dass ihre Methode besser als bisherige Modelle mit experimentellen und berechneten Referenzwerten übereinstimmt. Thallmair koordinierte die aufwendigen Arbeiten an Instituten in Frankreich, den Niederlanden, Spanien, Brasilien, Portugal, Polen und Südkorea. „Momentan arbeiten wir an einer weiteren Verbesserung des Proteinmodells, das eine noch realistischere Beschreibung der Proteinflexibilität erlaubt“, so Thallmair.
Publikation: Paulo C. T. Souza et. al., GōMartini 3: From large conformational changes in proteins to environmental bias corrections, Nat. Commun. 16, 4051, https://doi.org/10.1038/s41467-025-58719-0