May 9, 2025
Better description of large protein structures
More precise model represents flexibility and properties of proteins
A group led by FIAS Fellow Sebastian Thallmair is using a significantly improved method to optimize the calculation of protein structures. The international team has succeeded in describing the flexibility and mechanical properties of proteins more precisely.
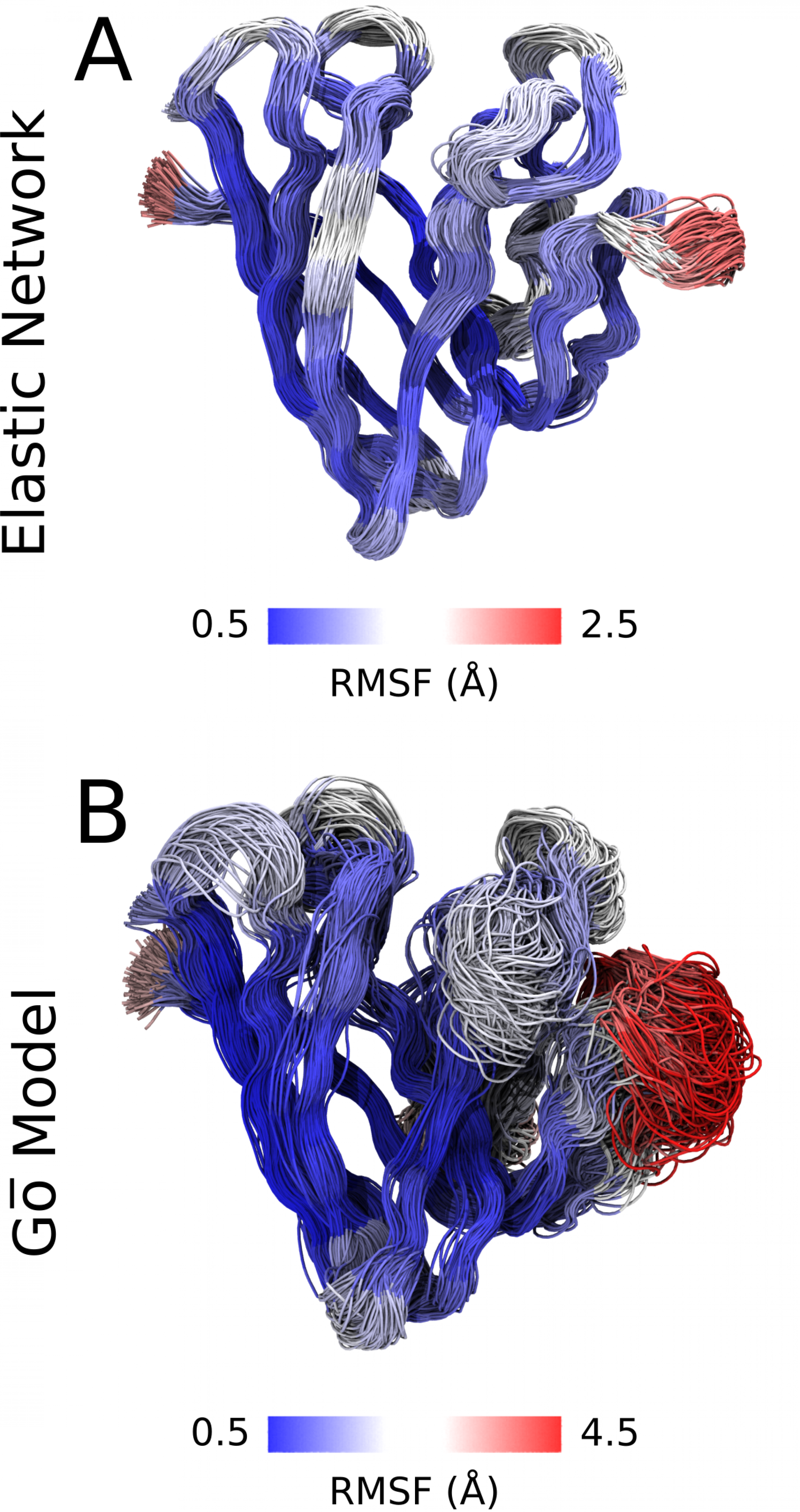
Calculating the structure of larger proteins or even protein complexes atom by atom is extremely complex and requires a great deal of computing power - “it can take years”, explains FIAS Fellow Sebastian Thallmair. A large international team has now succeeded in improving these calculations. They use a method that also reduces the computational effort. Instead of representing each atom individually, they combine them into small groups. This is crucial for a reduced computational effort. However, since “smaller” interactions such as hydrogen bonds decisively stabilize protein structures, these must be partially reintroduced. The team has developed a significantly improved method that stabilizes protein models while allowing them a high degree of flexibility.
“By grouping atoms, we calculate with one-tenth to one-twentieth of the particles and only have to consider one hundred to one thousand additional bonds in our optimized model in order to realistically describe the flexibility of the proteins,” explains Thallmair. “In addition, the versatile method also allows us to refine the interaction with the environment.” The model is therefore significantly more efficient than calculations of proteins with atomistic resolution and, thanks to improvements, can be used on appropriate supercomputers such as those available at FIAS. The widely used Martini model for calculating proteins therefore gets more easily applicable and also reproduces protein dynamics much better.
Thallmair and his team use various examples to demonstrate these optimizations:
- Using proteins that perform important mediation tasks between inside and outside in the membrane of cells, they show that their model represents their structure at least comparably well and at the same time better describes the elasticity of the proteins - their mobility depending on external influences.
- They show the same effect for proteins that bind other molecules, such as drugs: The improved flexibility of proteins makes it possible, for example, to describe more precisely how protein pockets adapt when a molecule binds.
- The improved mechanical properties are evident in a protein whose mutation in just one amino acid causes a serious disease of the nervous system (amyotrophic lateral sclerosis). The change leads to the active center opening up further, causing the protein to lose its function. It was crucial for this observation that the researchers were able to achieve a simulation time of one millisecond thanks to the reduced computational effort.
- Using mutations in the spike protein of the coronavirus, the researchers showed that the method can also describe how the mutations improve the interactions between host and virus.
With the wide range of examples, the team shows that their method agrees better than previous models with experimental and calculated reference values. Thallmair coordinated the extensive work at institutes in France, the Netherlands, Spain, Brazil, Portugal, Poland, and South Korea. “We are currently working on further improving the protein model, which will allow an even more realistic description of protein flexibility,” says Thallmair.
Publication: Paulo C. T. Souza et. al., GōMartini 3: From large conformational changes in proteins to environmental bias corrections, Nat. Commun. 16, 4051, https://doi.org/10.1038/s41467-025-58719-0